
Modelling and simulation of crystal-amorphous interfaces in semi-crystalline polymers
Project description:
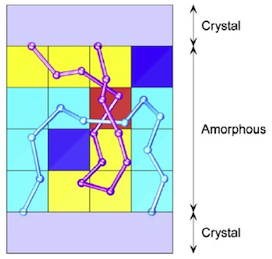
The purpose of this project is to further develop a recently generated Monte Carlo simulation by invoking information obtained by new experiments (primarily synchrotron small-angle X-ray scattering and atomic force microscopy), to generate a general methodology applicable to semi-crystalline polymers different in repeating unit and molecular architecture in order to gain fundamental knowledge about the topological aspects of crystal-amorphous interfaces.
The final aims are as follows:
- Developed generic methodology including algorithms, computer code and specific experimental input data useful to predict the structure of crystal-amorphous interfaces with a special emphasis on the topological aspects (tie chains and trapped entanglements)
- Mapped and interpreted effects of repeating unit structure, molar mass, molecular architecture (short-chain branching, long-chain branching, star-branching and crosslinking) and crystallization conditions on the structure of the crystal-amorphous interface with an emphasis on the topological aspects